

NeurIPS Paper by Asst. Prof. Eric Jonas Delegates Spectroscopy to the Machines
 It’s a rite of passage for every organic chemistry student: learning the difficult, laborious process of decoding spectroscopy data. For over 150 years, scientists and students have squinted at the peaks and valleys produced by these techniques in order to determine the molecular structure of a mystery sample. And in the computer age, many researchers have attempted to automate this “molecular fingerprint” analysis, with only limited success.
It’s a rite of passage for every organic chemistry student: learning the difficult, laborious process of decoding spectroscopy data. For over 150 years, scientists and students have squinted at the peaks and valleys produced by these techniques in order to determine the molecular structure of a mystery sample. And in the computer age, many researchers have attempted to automate this “molecular fingerprint” analysis, with only limited success.
But recent advances in machine learning, simulation, graph theory, and other computational approaches may have finally paved the way for automation of this chemistry lab linchpin. In a paper presented at the 2019 NeurIPS meeting, UChicago CS Assistant Professor Eric Jonas described a new technique for reading nuclear magnetic resonance (NMR) spectra, opening up new possibilities for chemical analysis and the design of new molecules using a “self-driving spectrometer.”
“I think in the future, the ability to hand-read spectra will be much less important,” Jonas said. “It’s such a natural fit for machine learning and such an important area for science.”
Jonas’ paper, “Deep imitation learning for molecular inverse problems,” tackles what’s typically a one-way street between molecules and their spectra. If you know a given molecule’s structure, scientists (or computers) can very accurately predict the spectroscopy results a measurement will produce. But the reverse situation, where a scientist is handed spectroscopy data and asked to deduce the original structure, is far more difficult and time-consuming.
“When you make a spectroscopic measurement of a molecule, it’s going to tell you lots of things about, say, the bonds in that molecule or the local electronic environment of a particular nucleus,” Jonas said. “You get a lot of these different pieces of information, but putting those pieces of information back together is of course the hard part. That’s what chemists spend a lot of their day doing.”
But those same frustrations make reading spectroscopy data a natural candidate for machine learning. In the same way that an image classification neural network learns its own “rules” for determining whether a photograph is a dog or a cat, a similar model could figure out how to interpret the features of a spectra and make an educated guess about the structure it encodes. The problem, as with many machine learning applications, is finding enough data to train the model; for nuclear magnetic resonance spectroscopy, accurately measuring just one sample can take minutes to hours.
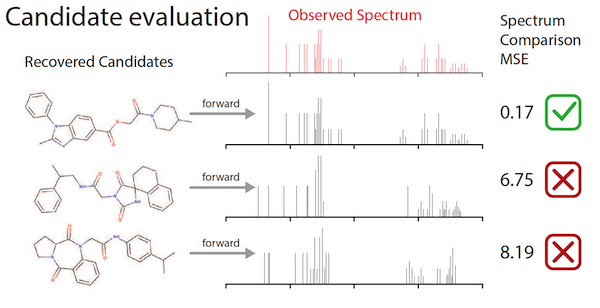
To remedy this shortage, Jonas and colleagues at De Montfort University in the UK first created a “fast-forward model” that created synthetic spectra data for over one million molecules. He then used that data to train a deep neural network, drawing upon imitation learning — a variant of reinforcement learning — to improve its accuracy in assembling molecular structures that fit the data input.
“There’s a very close mapping between the kind of problems that we think about for machine learning for graphs and the way we think about these kind of spectroscopic measurements,” Jonas said. “Part of our insight was recognizing that sequentially building a molecule where you have a known true answer can be treated as an imitation learning problem. I can show the model a tremendously large number of examples where I say, here’s the partially assembled molecule, and here’s the list of correct next edges. And it just has to learn how to go from a partially-completed molecule and observed spectral data to the right next edge.”
The novelty of that approach earned Jonas and the paper a spot at the 2019 Neural Information Processing Systems (NeurIPS) conference, one of the leading gatherings for machine learning research. But with some additional refinements, he hopes the technique will most significantly impact chemistry and other biological and physical sciences.
Currently, the model only works on molecules made up of the four most common elements: carbon, hydrogen, oxygen, and nitrogen. But a system that can accurately and rapidly identify even small molecules could dramatically accelerate work in microbiome research, drug discovery, and other areas in perpetual search of new chemical structures. Automating the analysis of spectra could also open up a more active discovery process, where spectrometers not only provide measurements but also suggestions of where to look next in the pursuit of molecules with useful properties.
“If we can develop algorithms that are good enough to understand the spectra, then we can start developing algorithms that are good enough to figure out the right next measurements to make,” Jonas said. “We’re all very excited about the direction that active learning can potentially take us for this kind of scientific instrumentation.”










